Open File Formats

Open standards in the realm of analytical chemistry have existed for a long time. Some have better marketing than others. Time for an overview of what already exists.
JCAMP-DX

The earliest open standard that is still around is JCAMP-DX from the International Union of Pure and Applied Chemistry, which in the world of chemistry is the authoritative source. The JCAMP working group focused on spectroscopy and spectrometry. Chromatography was also addressed but published only as a final draft. An example file looks like this:
##TITLE= Headspace from ripe banana
##JCAMP-DX= 4.24
##DATA TYPE= GAS CHROMATOGRAPH
##ORIGIN= HP 5890 GC TABULATED file
##OWNER= Dept of Chem, UWI, Mona, JAMAICA
##DATE= 11-26-1995
##TIME= 08:51:07
##NPOINTS= 6003
##XUNITS= TIME
##YUNITS= ARBITRARY
##FIRSTX= 0.00000
##LASTX= 20.00700
##XFACTOR= 1.0
##YFACTOR= 1.0
##FIRSTY= 48925
##MAXY= 128636
##MINY= 48310
##XYDATA= (X++(Y..Y))
0.00000 48925 48918 48909 48922 48912
0.01700 48916 48922 48920 48913 48920
0.03300 48921 48914 48910 48924 48928
[...]I believe the formatting was chosen to be backwards compatible with 80-column displays, needle printers, postal services, and telefax. Remember, these were pioneers when personal computing was still in its infancy. You can edit it with a text editor, but only to some degree. JCAMP-DX supports multiple ways of storing and compressing data. The example above uses delta compression for the x-axis for example, as GC typically has a constant scan rate.
Even though it is text, you clearly need a tool to plot it. You can read these legacy files in OpenChrom just fine.
##TITLE= diff
##JCAMPDX= 5.0 $$ Bruker NMR JCAMP-DX V1.0
##DATA TYPE= NMR Spectrum
##DATA CLASS= XYDATA
##ORIGIN= uk
##OWNER= uk
##.OBSERVE FREQUENCY= 100.4
##.OBSERVE NUCLEUS= ^13C
##.ACQUISITION MODE= SIMULTANEOUS
##.AVERAGES= 32
##.DIGITISER RES= 16
##SPECTROMETER/DATA SYSTEM= JEOL GX 400
$$ Bruker specific parameters
$$ --------------------------
##$AQ_mod= 1
##$AUNM= <au_zgsino>
##$BF1= 100.4
##$BF2= 360.131842
##$BF3= 100.13
##$BF4= 500.13
##$BYTORDA= 0
##$CNST= (0..31)
[...]If you look at this file from Bruker for NMR, you can see adoption with plenty of vendor-specific fields that are still somewhat relevant up to today.
OpenChrom can read those as well. Despite its name, it actually has support for NMR data and basic processing built in.
##TITLE= FIX form (FILE: jtpolys.jdx)
##JCAMP-DX= 4.24 $$ Software version unknown
##DATA TYPE= INFRARED SPECTRUM
##ORIGIN= JCAMP-DX Test Disk 1.04
##OWNER= Public Domain
##$URL= http://wwwchem.uwimona.edu.jm:1104/spectra/testdata/index.html
##SPECTROMETER/DATA SYSTEM= Digilab Data system=3240
##XUNITS= 1/CM
##YUNITS= TRANSMITTANCE
##RESOLUTION= 4.0
##FIRSTX= 4.47484259e+02
##LASTX= 4.00228378e+03
##FIRSTY= 9.816334969e-01
##MAXY= 1.022816066
##MINY= 3.428528714e-01
##XFACTOR= 1.92881146e+00
##YFACTOR= 2.384185791e-09 $$ this expands data to fill a 32 bit word
##NPOINTS= 1844
##XYDATA= (X++(Y..Y))
232 411726930 412183219 411759721 414612851 419274055 419812369 417860195
239 420451535 420858525 418370372 418228023 418099321 419213585 423393589
246 425833398 424330751 423232014 425027331 425868292 425158533 425993283The same is true for FT-IR, which is supported out of the box in OpenChrom in read-only mode.
UV-Vis is another technique JCAMP-DX was initially designed for. It can also be read by OpenChrom to provide backwards compatibility with old data, although we usually aim to support the vendor formats directly.
##TITLE= pepsi-cola (diluted)
##JCAMP-DX= 4.24
##DATA TYPE= UV/VIS SPECTRUM
##ORIGIN= Dept of Chemistry, UWI, JAMAICA
##OWNER= public domain
##DATE= 97/06/23
##TIME= 09:55:44.00
##SPECTROMETER/DATA SYSTEM= PERKIN-ELMER LAMBDA 19 UV/VIS/NIR UV
##RESOLUTION= 1.000000
##DELTAX= -1.000000
##XUNITS= nm
##YUNITS= A
##XFACTOR= 1.00
##YFACTOR= .00000011920928955078
##FIRSTX= 320.00
##LASTX= 220.00
##NPOINTS= 101
##FIRSTY= 3.95
##MAXY= 1.033029
##MINY= 3.95
##XYDATA= (X++(Y..Y))
320.000000 8665676 8782337 8902319 9027385 9154514
315.000000 9287029 9423184 9562309 9706719 9854241
310.000000 10002015 10154503 10305985 10460863 10613729
[...]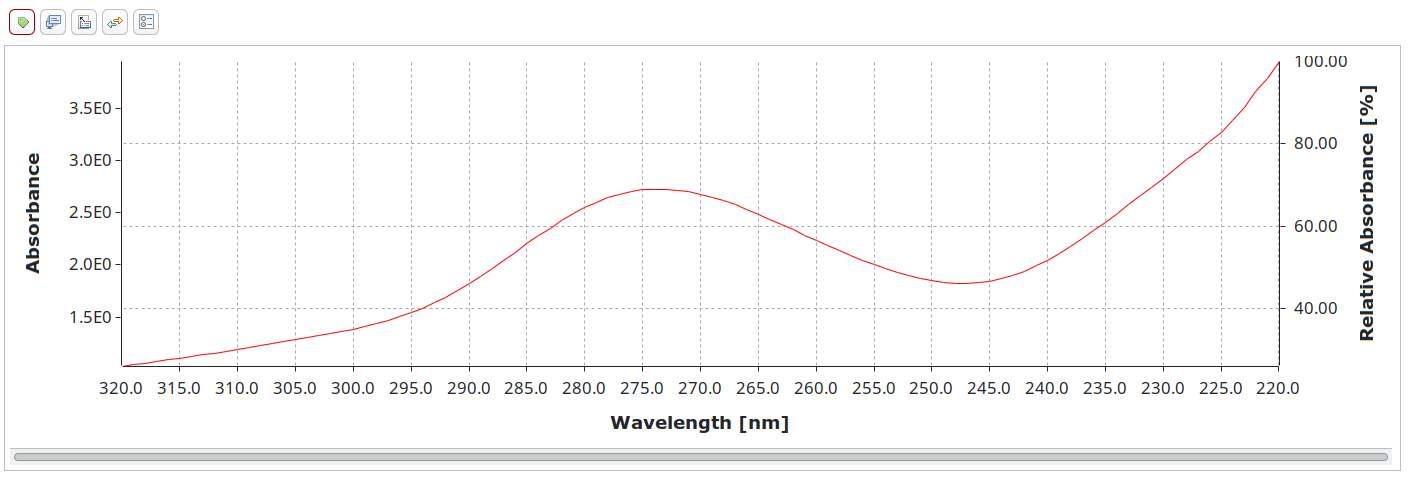
Did it ever take off? In a postmortem, the authors of JCAMP-DX complained that despite best efforts, vendors deviated from the specifications, causing incompatibility problems.
CML
You probably know HTML or have seen it when accidentally looking at the source code of a website. The Chemical Markup Language is an early attempt to put structured data in XML. It is structured, still somewhat human-readable, and can be validated against a schema. It does not seem to feature compression yet.
<?xml version="1.0" standalone="yes"?>
<spectrum id="sp04" type="UV/VIS"
xmlns="http://www.xml-cml.org/schema"
xsi:schemaLocation="http://www.xml-cml.org/schema ../../schema.xsd"
xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance"
>
<metadataList>
<metadata name="dc:origin">INSTITUTE OF ENERGY PROBLEMS OF CHEMICAL PHYSICS, RAS</metadata>
<metadata name="dc:owner">INEP CP RAS, NIST OSRD</metadata>
</metadataList>
<sample>
<molecule>
<name convention="cas:regno">109-99-9</name>
</molecule>
</sample>
<spectrumData>
<xaxis>
<array dataType="xsd:double" units="unit:nm">
[...]
162.6391 162.6163 162.5935 162.5382 162.4822 162.4249 162.3758 162.3188 161.9523 </array>
</xaxis>
<yaxis multiplierToData="1.000000">
<array dataType="xsd:double" units="unit:unknown">
2.101893 2.101893 2.103183 2.103183 2.104424 2.104424 2.105568 2.023428 2.029936
[...]
</yaxis>
</spectrumData>
</spectrum>
Did it take off? Probably not in analytical chemistry, but it is still being supported as an exchange format for molecular structures despite proprietary quasi-standards dominating the field.
Analytical Data Interchange Format
The Analytical Data Interchange Format, short ANDI, or just .cdf from its file extension, is a binary format, but an open one. It uses NetCDF as a container and a specification on how to store chromatograms. As an ASTM method, they of course had to verify it. They did a ring trial with all the participating vendors to validate compatibility. A major downside is that files can become larger than binary vendor formats. It is still somewhat relevant today because of software support and being the only published ASTM standard in the field.
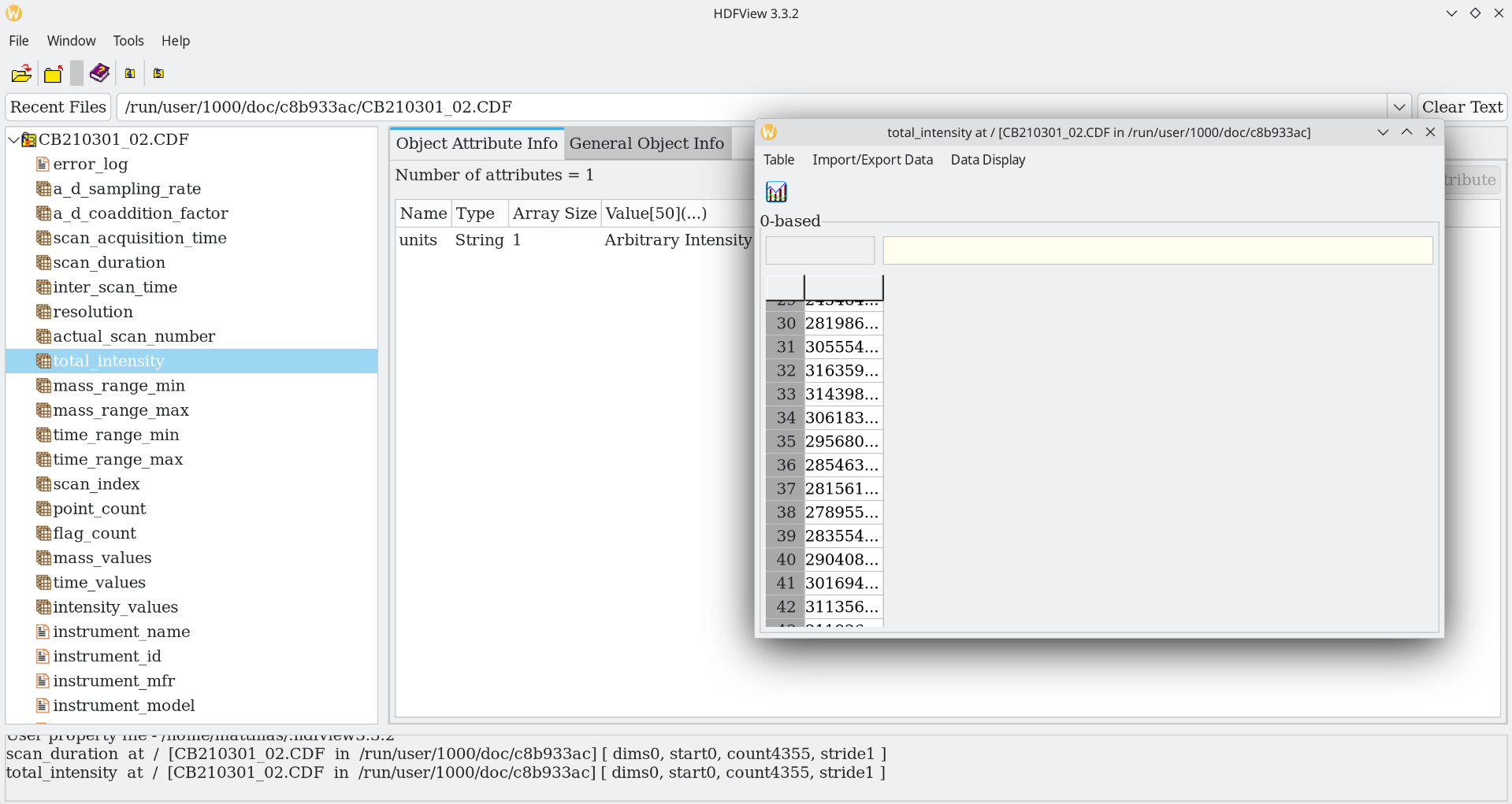
As netCDF is a self-describing open container format, you can inspect these files with generic readers such as HDFView from HDF group.
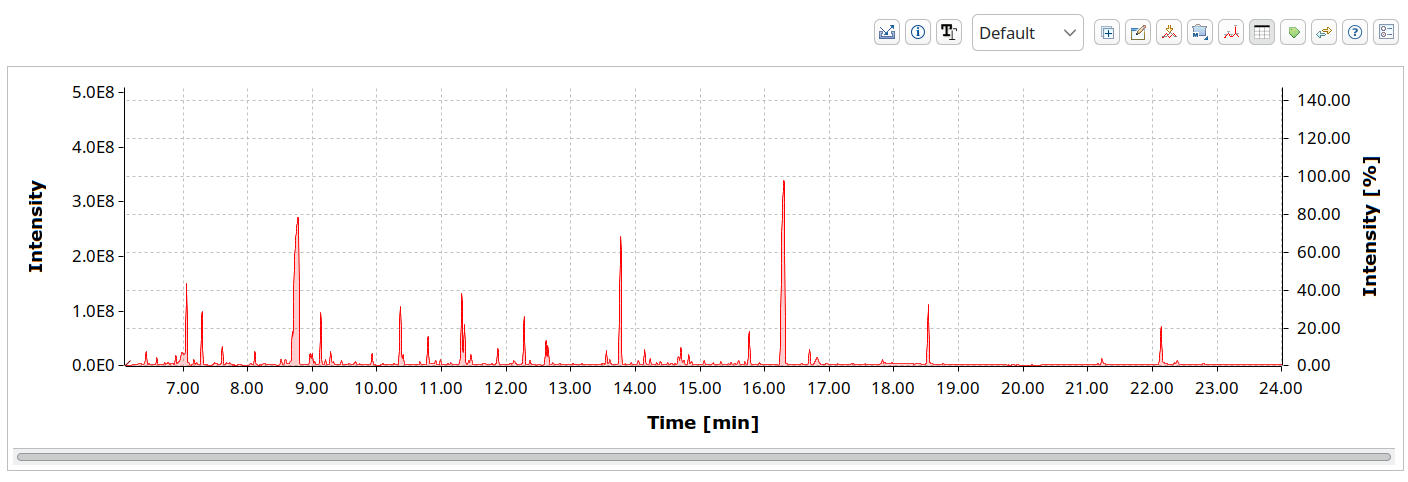
OpenChrom can read and write CDF files. It is used to communicate with older software such as AMDIS.
Generalized Analytical Markup Language
Generalized Analytical Markup Language (short GAML) is another XML-based format with simple X/Y and non-standardized metadata. The schemas are public domain, but I'd still call this a vendor format because it was envisioned by Thermo Fisher Scientific to consolidate file formats after company takeovers, was meant to be an export format into a Thermo LIMS and to comply with FDA and GDP regulatory guidelines for long term readability. However, most fields are actually a non-standardized dump of values that only have a meaning to developers within Thermo Fisher and differ by the software used to export.
<?xml version="1.0" encoding="UTF-8"?>
<GAML version="1.00" name="GASOLINE">
<integrity algorithm="SHA1">c64dbcd6b90d327e745bde107056c0cf1eebcfbc</integrity>
<parameter name="component_name" label="Component name" group="GAML Generation">GAMLIO</parameter>
<parameter name="component_version" label="Component version" group="GAML Generation">9.1.3.10</parameter>
<parameter name="FH_CDATE" label="Date created" group="Sequence File Header">2/22/1989</parameter>
[...]
<experiment name="gasoline">
<collectdate>1989-02-22T13:39:41Z</collectdate>
<trace name="Channel A Chromatogram" technique="CHROM">
<Xdata label="Minutes" units="MINUTES" valueorder="EVEN">
<values byteorder="INTEL" format="FLOAT64" numvalues="2912">
[...]
</values>
<Ydata units="UNKNOWN">
<values byteorder="INTEL" format="FLOAT32" numvalues="2912">
[...]
</values>
<peaktable>
[...]
<peak name="C15" number="83">
<peakXvalue>117.599450360701</peakXvalue>
<peakYvalue>11890</peakYvalue>
<baseline>
<startXvalue>116.677430436276</startXvalue>
<startYvalue>8776</startYvalue>
<endXvalue>118.060460322913</endXvalue>
<endYvalue>10943</endYvalue>
</baseline>
</peak>
[...]
</peaktable>
</Ydata>
</Xdata>
</trace>
</experiment>
</GAML>You can see that it exports not just raw data but also a peak table, which is pretty handy.
OpenChrom can read GAML files, as reading in large data lakes and assisting in the migration to modern platforms is what we offer to customers.
AnIML

Analytical Information Markup Language, mostly known for its abbreviation, was envisioned around the millennium and is an upcoming ASTM standard that is still a draft today. It is a construction kit for analytical data. A core schema defines data structures, as in how numbers are stored and how techniques are defined. The techniques are modular building blocks. For example, chromatography plus mass spectrometry combines into something that can be used for GC-MS.
<?xml version="1.0" encoding="UTF-8" standalone="yes"?>
<AnIML xmlns:ns2="http://www.w3.org/2000/09/xmldsig#"
xmlns:ns3="urn:org:astm:animl:schema:core:draft:0.90" version="0.90">
<SampleSet>
<Sample sampleID="DemoChromatogram8137928444050239328" barcode="" comment="" name="" />
</SampleSet>
<ExperimentStepSet>
<ExperimentStep experimentStepID="1" name="Q1">
<Technique uri="https://animl.openchrom.net/chromatography.atdd" name="Chromatography" />
<Method>
<Author>
<Name>Matt</Name>
</Author>
<Software>
<Manufacturer>Lablicate GmbH</Manufacturer>
<Name>OpenChrom</Name>
<Version>1.5.31.qualifier</Version>
<OperatingSystem>Linux</OperatingSystem>
</Software>
</Method>
<Result name="Separation Monitoring">
<SeriesSet length="0" name="Separation Monitoring">
<Series dependency="independent" plotScale="linear" seriesType="Float32" name="Time"
id="timeSeries">
<EncodedValueSet></EncodedValueSet>
<Unit label="ms" quantity="Time">
<SIUnit exponent="-3.0">s</SIUnit>
</Unit>
</Series>
</SeriesSet>
<ExperimentStepSet>
<ExperimentStep sourceDataLocation="/tmp/DemoChromatogram8137928444050239328.ocb"
name="Total Signal">
<Technique uri="https://animl.openchrom.net/ms-trace.atdd" name="Mass Spectrum Time Trace" />
<Result>
<SeriesSet length="5726" name="Mass Chromatogram" id="TIC">
<Series dependency="independent" plotScale="linear" seriesType="Int32"
name="Retention Time"></Series>
<Unit label="Abundance" quantity="arbitrary" />
</Series>
</SeriesSet>
<Category>
<Parameter>
<S>TIC</S>
</Parameter>
</Category>
</Result>
</ExperimentStep>
<ExperimentStep experimentStepID="ms-1" name="MS #1">
<Technique uri="https://animl.openchrom.net/mass-spec.atdd" name="Mass Spectrometry" />
<Result name="Spectrum">
<SeriesSet length="35" name="Spectrum">
<Series dependency="dependent" plotScale="linear" seriesType="Float64"
name="Mass/Charge">
<EncodedValueSet>[...]</EncodedValueSet>
<Unit label="Mass/Charge Ratio" quantity="mz" />
</Series>
<Series dependency="dependent" plotScale="linear" seriesType="Float32" name="Intensity">
<EncodedValueSet>[...]</EncodedValueSet>
<Unit label="Abundance" quantity="arbitrary" />
</Series>
</SeriesSet>
</Result>
[...]
</ExperimentStep>
The caveat with AnIML is that ASTM is about to only standardize the core schema, none of the techniques, and even that process seems stuck. In fact, most of the techniques have never been published and stay a company secret. Whether this is truly open or a standard is at least debatable. On the plus side, it gives the vendors more flexibility without the need to negotiate consensus first. There is no validator and no example data available to the public. You can use AnIML Viewer as a substitute. Even if you are just mostly serializing to non-canonical XML for internal usage, that is still better than nothing.
OpenChrom can read/write a selection of the published techniques that are available in a volatile source code repository without release management, so compatibility will be a challenge.
mzML
mzXML and mzData are both initiatives to save MS and LC-MS data as XML from the proteomics community. As both initiatives noticed that they were competing, and now everyone has to implement two standards rather than one, they joined forces... to create a new format called mzML that merges the ontological and the more strict schema adherence approach of both now deprecated predecessors. It stores the data values in binary form with optional ZIP compression.
<?xml version="1.0" encoding="utf-8"?>
<mzML xmlns="http://psi.hupo.org/ms/mzml" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xsi:schemaLocation="http://psi.hupo.org/ms/mzml http://psidev.info/files/ms/mzML/xsd/mzML1.1.0.xsd" id="20190702_Kristine01_museumsamples3_A17" version="1.1.0">
<cvList count="2">
<cv id="MS" fullName="Proteomics Standards Initiative Mass Spectrometry Ontology" version="3.44.0" URI="http://psidev.cvs.sourceforge.net/*checkout*/psidev/psi/psi-ms/mzML/controlledVocabulary/psi-ms.obo"/>
<cv id="UO" fullName="Unit Ontology" version="12:10:2012" URI="http://obo.cvs.sourceforge.net/*checkout*/obo/obo/ontology/phenotype/unit.obo"/>
</cvList>
<fileDescription>
<fileContent>
<cvParam cvRef="MS" accession="MS:1000579" name="MS1 spectrum"/>
<userParam name="MALDIquantForeign" value="MALDIquant object(s) exported to mzML"/>
</fileContent>
<sourceFileList count="1">
<sourceFile id="SF1" location="/Users/kkrichter/Google Drive/Old/Jena_ZooMS/MALDI-Spctra/2019-07-02-Kristine01/20190702_Kristine01_museumsamples3/0_A17/1/1SRef" name="fid">
<cvParam cvRef="MS" accession="MS:1000825" name="Bruker FID file"/>
<cvParam cvRef="MS" accession="MS:1000773" name="Bruker FID nativeID format"/>
<cvParam cvRef="MS" accession="MS:1000569" name="SHA-1" value="503c863d73f889d10d4e2d41c8ec1fc1f39c3c53"/>
</sourceFile>
</sourceFileList>
</fileDescription>
<softwareList count="1">
<software id="MALDIquantForeign" version="0.12"/>
</softwareList>
<instrumentConfigurationList count="1">
<instrumentConfiguration id="IC0"/>
</instrumentConfigurationList>
<dataProcessingList count="1">
<dataProcessing id="export">
<processingMethod order="1" softwareRef="MALDIquantForeign">
<userParam name="MALDIquant object(s) exported to mzML" value=""/>
</processingMethod>
</dataProcessing>
</dataProcessingList>
<run id="run0" defaultInstrumentConfigurationRef="IC0">
<spectrumList count="1" defaultDataProcessingRef="export">
<spectrum index="0" id="da47942e-fdf3-4cb3-88fa-6c696623b019" defaultArrayLength="176981" spotID="20190702_Kristine01_museumsamples3.A17">
<cvParam cvRef="MS" accession="MS:1000511" name="ms level" value="1"/>
<cvParam cvRef="MS" accession="MS:1000294" name="mass spectrum"/>
<cvParam cvRef="MS" accession="MS:1000528" name="lowest observed m/z" value="799.729867312381" unitCvRef="MS" unitAccession="MS:1000040" unitName="m/z"/>
<cvParam cvRef="MS" accession="MS:1000527" name="highest observed m/z" value="3496.45919544856" unitCvRef="MS" unitAccession="MS:1000040" unitName="m/z"/>
<cvParam cvRef="MS" accession="MS:1000128" name="profile spectrum"/>
<cvParam cvRef="MS" accession="MS:1000285" name="total ion current" value="153562.949977686"/>
<binaryDataArrayList count="2">
<binaryDataArray encodedLength="1398316">
<cvParam cvRef="MS" accession="MS:1000574" name="zlib compression"/>
<cvParam cvRef="MS" accession="MS:1000523" name="64-bit float"/>
<cvParam cvRef="MS" accession="MS:1000514" name="m/z array" unitCvRef="MS" unitAccession="MS:1000040" unitName="m/z"/>
<binary>[..]</binary>
</binaryDataArray>
<binaryDataArray encodedLength="294368">
<cvParam cvRef="MS" accession="MS:1000574" name="zlib compression"/>
<cvParam cvRef="MS" accession="MS:1000523" name="64-bit float"/>
<cvParam cvRef="MS" accession="MS:1000515" name="intensity array" unitCvRef="MS" unitAccession="MS:1000131" unitName="number of counts"/>
<binary>[...]</binary>
</binaryDataArray>
</binaryDataArrayList>
</spectrum>
</spectrumList>
</run>
</mzML>
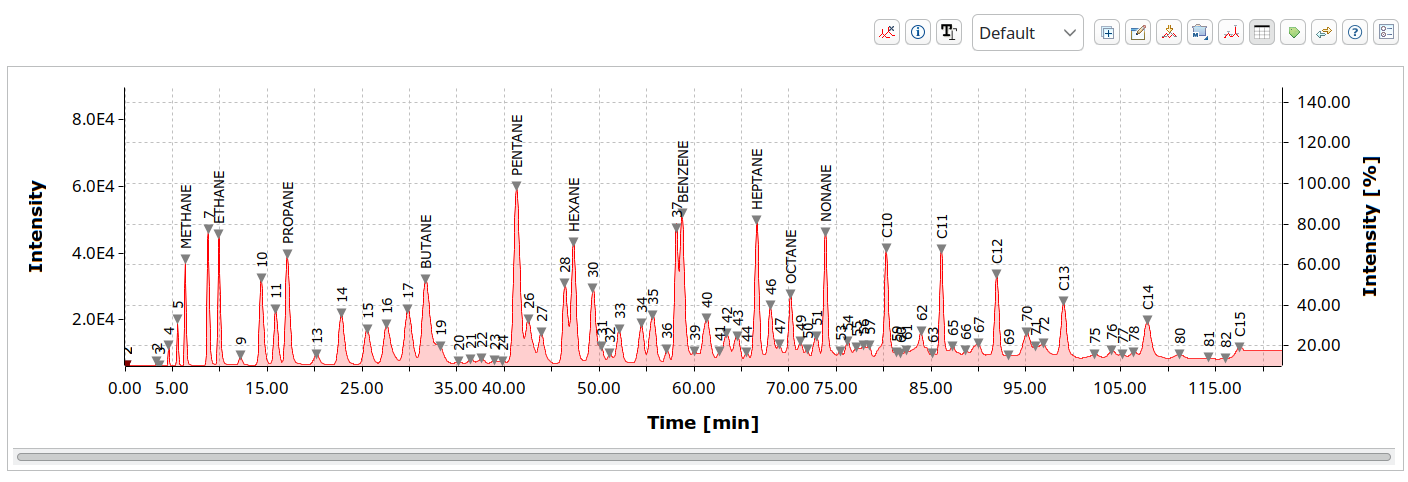
Both single mass spectra for techniques such as MALDI-TOF MS
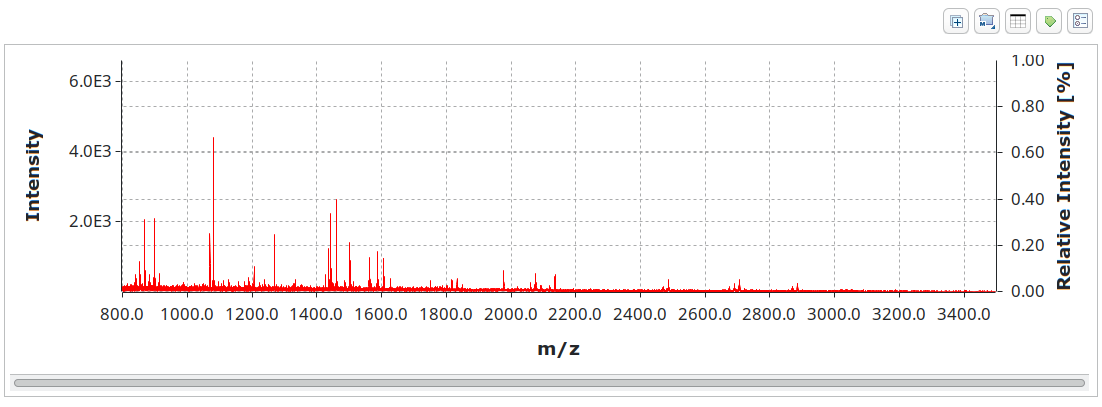
and hyphenated with chromatography like LC-MS or MS/MS are supported.
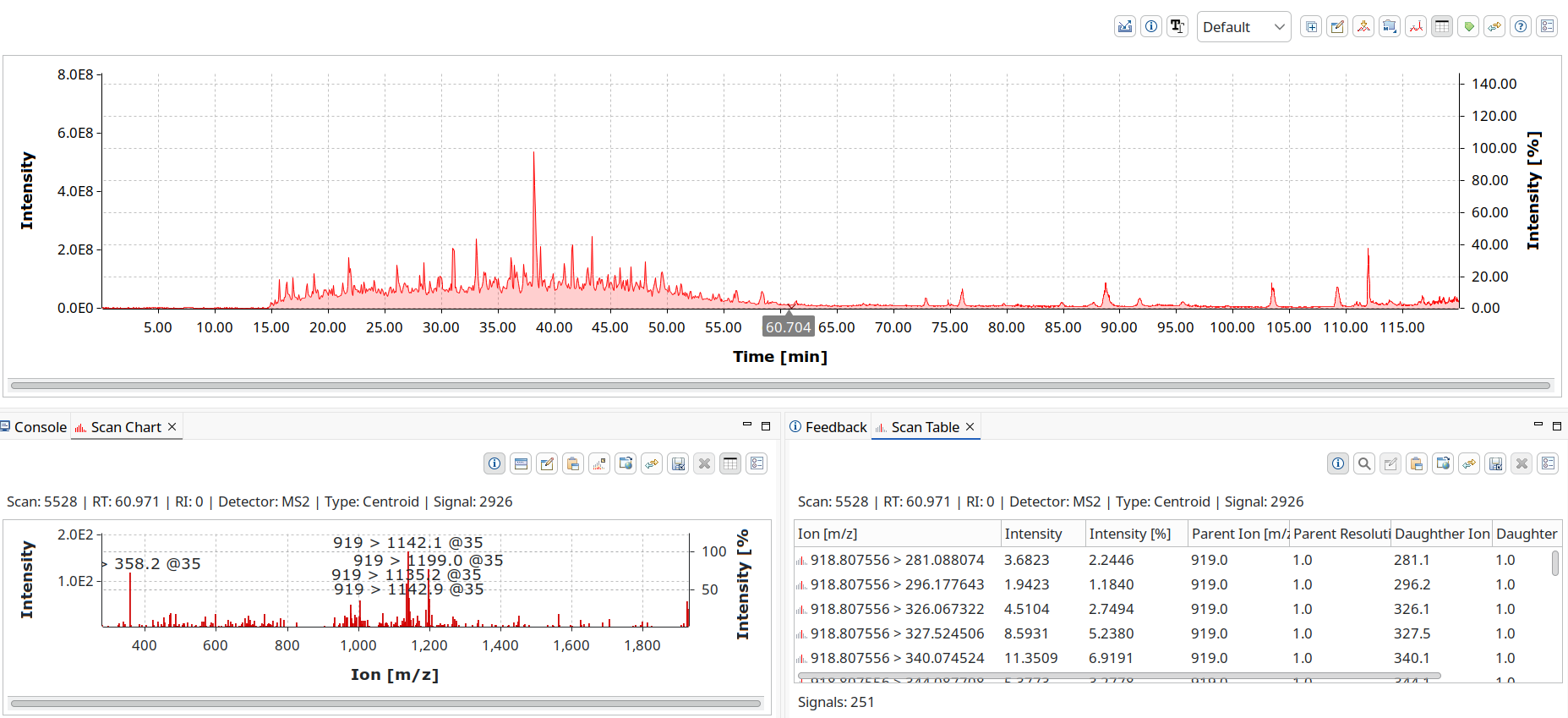
mzML is widely used in proteomics toolchains. OpenChrom has read/write support that we built together with developers from downstream programs for best compatibility and read speeds.
nmrML

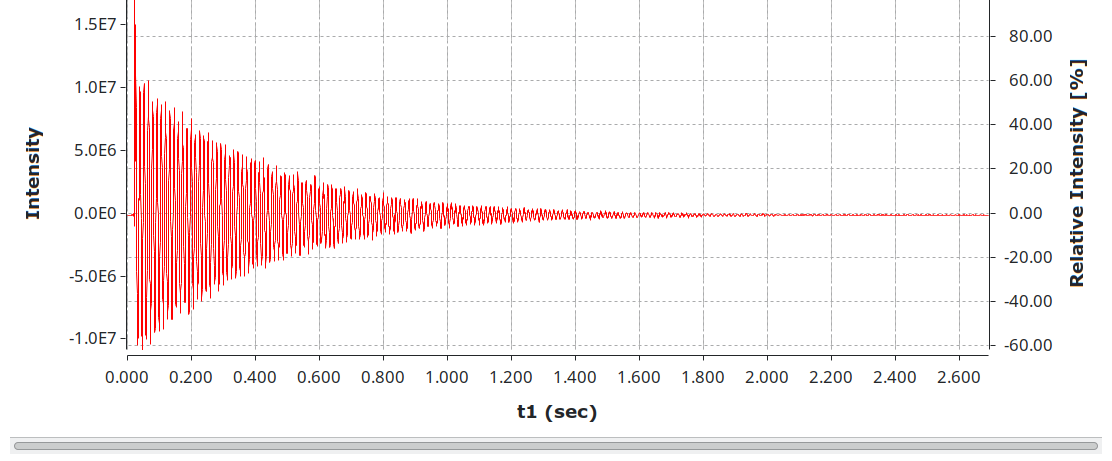
I bet you can guess what this is already, as there is a clear pattern now established. nmrML is an extensible markup language for nuclear magnetic resonance spectroscopy. Clearly inspired by the mzML format, it also makes use of a controlled vocabulary to store metadata and a strict format for data storage. It is easier to read and validate than JCAMP-DX.
<?xml version="1.0" encoding="UTF-8" standalone="yes"?>
<nmrML xmlns="http://nmrml.org/schema" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" version="1.0.rc1" xsi:schemaLocation="http://nmrml.org/schema http://nmrml.org/schema/v1.0.rc1/nmrML.xsd">
<cvList>
<cv id="NMRCV" fullName="Nuclear Magnetic Resonance CV" version="1.1.0" URI="http://nmrml.org/cv/v1.1.0/nmrCV.owl"/>
<cv id="UO" fullName="Unit Ontology" version="3.2.0" URI="http://purl.obolibrary.org/obo/"/>
<cv id="CHEBI" fullName="Chemical Entities of Biological Interest Ontology" version="105" URI="http://purl.obolibrary.org/obo/"/>
<cv id="NCIThesaurus" fullName="NCI Thesaurus" version="" URI="http://ncicb.nci.nih.gov/xml/owl/EVS/Thesaurus.owl#"/>
</cvList>
<fileDescription>
<fileContent>
<cvParam cvRef="NMRCV" accession="NMR:1400165" name="1D NMR acquisition parameter set"/>
</fileContent>
</fileDescription>
<contactList>
<contact id="ID00001" fullname="mica" email="mica@mhpbf141"/>
</contactList>
<sourceFileList>
<sourceFile id="ID00002" name="fid" location="file:/C:/Workdir/COSMOS/nmrML/examples/reference_spectra_examples/MMBBI/./MMBBI_10M12-CE01-1a/1/fid" sha1="f0083d8c6b161b088c5fea081ebf5e33ddf2c5bf">
<cvParam cvRef="NMRCV" accession="NMR:1400320" name="Bruker UXNMR/XWIN-NMR format"/>
<cvParam cvRef="NMRCV" accession="NMR:1400119" name="FID file"/>
</sourceFile>
<sourceFile id="ID00003" name="pulseprogram" location="file:/C:/Workdir/COSMOS/nmrML/examples/reference_spectra_examples/MMBBI/./MMBBI_10M12-CE01-1a/1/pulseprogram">
<cvParam cvRef="NMRCV" accession="NMR:1400320" name="Bruker UXNMR/XWIN-NMR format"/>
<cvParam cvRef="NMRCV" accession="NMR:1400122" name="pulse sequence file"/>
</sourceFile>
<sourceFile id="ID00004" name="acqus" location="file:/C:/Workdir/COSMOS/nmrML/examples/reference_spectra_examples/MMBBI/./MMBBI_10M12-CE01-1a/1/acqus">
<cvParam cvRef="NMRCV" accession="NMR:1400320" name="Bruker UXNMR/XWIN-NMR format"/>
<cvParam cvRef="NMRCV" accession="NMR:1002006" name="acquisition parameter file"/>
</sourceFile>
<sourceFile id="ID00101" name="procs" location="file:/C:/Workdir/COSMOS/nmrML/examples/reference_spectra_examples/MMBBI/./MMBBI_10M12-CE01-1a/1/pdata/1/procs">
<cvParam cvRef="NMRCV" accession="NMR:1400320" name="Bruker UXNMR/XWIN-NMR format"/>
<cvParam cvRef="NMRCV" accession="NMR:1400123" name="processing parameter file"/>
</sourceFile>
<sourceFile id="ID00102" name="1r" location="file:/C:/Workdir/COSMOS/nmrML/examples/reference_spectra_examples/MMBBI/./MMBBI_10M12-CE01-1a/1/pdata/1/1r" sha1="e118a59a206281a13a2ef2a33cadcfa468438951">
<cvParam cvRef="NMRCV" accession="NMR:1400320" name="Bruker UXNMR/XWIN-NMR format"/>
<cvParam cvRef="NMRCV" accession="NMR:1000319" name="1R file"/>
</sourceFile>
</sourceFileList>
<softwareList>
<software id="ID00005" version="Version 2.1" cvRef="NMRCV" accession="NMR:1400215" name="Bruker TopSpin software"/>
<software id="ID00103" version="Version 3.0" cvRef="NMRCV" accession="NMR:1400215" name="Bruker TopSpin software"/>
</softwareList>
<instrumentConfigurationList>
<instrumentConfiguration id="ID00006">
<cvParam cvRef="NMRCV" accession="NMR:1400198" name="Bruker NMR instrument"/>
<userParam name="Instrument Name" value="spect"/>
<userParam name="ProbeHead" value="5 mm PABBI 1H/D-BB Z-GRD Z859201/0037"/>
<softwareRef ref="ID00005"/>
</instrumentConfiguration>
</instrumentConfigurationList>
<acquisition>
<acquisition1D>
<acquisitionParameterSet numberOfSteadyStateScans="0" numberOfScans="64">
<contactRefList>
<contactRef ref="ID00001"/>
</contactRefList>
<softwareRef ref="ID00005"/>
<sampleContainer cvRef="NMRCV" accession="NMR:1400132" name="NMR Sample tube"/>
<sampleAcquisitionTemperature value="300.00" unitAccession="UO_0000012" unitName="kelvin" unitCvRef="UO"/>
<spinningRate value="4200" unitAccession="UO_0000169" unitName="dimensionless" unitCvRef="UO"/>
<relaxationDelay value="25.000000000000" unitAccession="UO_0000010" unitName="second" unitCvRef="UO"/>
<pulseSequence>
<userParam name="Pulse Program" value="zg"/>
</pulseSequence>
<shapedPulseFile ref="ID00003"/>
<groupDelay value="76.0000" unitAccession="UO_0000169" unitName="dimensionless" unitCvRef="UO"/>
<acquisitionParameterRefList>
<acquisitionParameterFileRef ref="ID00002"/>
<acquisitionParameterFileRef ref="ID00003"/>
<acquisitionParameterFileRef ref="ID00004"/>
</acquisitionParameterRefList>
<DirectDimensionParameterSet decoupled="false" numberOfDataPoints="32768">
<acquisitionNucleus cvRef="CHEBI" accession="CHEBI_49637" name="hydrogen atom"/>
<effectiveExcitationField value="500.160000000000" unitAccession="UO_0000325" unitName="megaHertz" unitCvRef="UO"/>
<sweepWidth value="6002.400960384150" unitAccession="UO_0000106" unitName="hertz" unitCvRef="UO"/>
<pulseWidth value="10.430210000000" unitAccession="UO_0000029" unitName="microsecond" unitCvRef="UO"/>
<irradiationFrequency value="500.162500800000" unitAccession="UO_0000325" unitName="megaHertz" unitCvRef="UO"/>
<irradiationFrequencyOffset value="2500.800000000000" unitAccession="UO_0000325" unitName="megaHertz" unitCvRef="UO"/>
<decouplingNucleus cvRef="NMRCV" accession="NMR:1000055" name="off resonance decoupling"/>
<samplingStrategy cvRef="NMRCV" accession="NMR:1000349" name="uniform sampling"/>
</DirectDimensionParameterSet>
</acquisitionParameterSet>
<fidData compressed="true" encodedLength="135344" byteFormat="Complex128">[...]</acquisition1D>
</acquisition>
<spectrumList>
<spectrum1D numberOfDataPoints="32768" id="ID00104" name="1">
<processingSoftwareRefList>
<softwareRef ref="ID00103"/>
</processingSoftwareRefList>
<processingParameterFileRefList>
<processingParameterFileRef ref="ID00101"/>
<processingParameterFileRef ref="ID00102"/>
</processingParameterFileRefList>
<spectrumDataArray compressed="true" encodedLength="126820" byteFormat="float64">[...]</spectrumDataArray>
<xAxis unitAccession="UO_0000169" unitName="parts per million" unitCvRef="UO" startValue="11.099150" endValue="-0.901812"/>
<processingParameterSet>
<postAcquisitionSolventSuppressionMethod cvRef="NCIThesaurus" accession="C19377" name="Not Defined"/>
<calibrationCompound cvRef="NCIThesaurus" accession="C19377" name="Not Defined"/>
<dataTransformationMethod cvRef="NMRCV" accession="NMR:1000070" name="FID fourier transformation"/>
</processingParameterSet>
<firstDimensionProcessingParameterSet>
<zeroOrderPhaseCorrection value="72.823770" unitAccession="UO_0000185" unitName="degree" unitCvRef="UO"/>
<firstOrderPhaseCorrection value="0.000000" unitAccession="UO_0000185" unitName="degree" unitCvRef="UO"/>
<calibrationReferenceShift value="undefined" unitAccession="UO_0000169" unitName="dimensionless" unitCvRef="UO"/>
<spectralDenoisingMethod cvRef="NCIThesaurus" accession="C19377" name="Not Defined"/>
<windowFunction>
<windowFunctionMethod cvRef="NMRCV" accession="NMR:1400069" name="exponential multiplication window function"/>
<windowFunctionParameter cvRef="NMRCV" accession="NMR:1400097" name="Line Broadening" value="0.300000"/>
</windowFunction>
<baselineCorrectionMethod cvRef="NCIThesaurus" accession="C19377" name="Not Defined"/>
</firstDimensionProcessingParameterSet>
</spectrum1D>
</spectrumList>
</nmrML>
OpenChrom has basic read support for it.
RDML
Again, an XML-based standard. Real-Time PCR Data Markup Language shows that this concept can be applied to molecular biology, and people feel there is a need. It not only maps the data but also describes the experiment. The file is a zipped archive, and inside you can find an XML.
<rdml xmlns:rdml="http://www.rdml.org" xmlns="http://www.rdml.org" version="1.3">
<dateMade>2011-01-09T17:04:55</dateMade>
<dateUpdated>2021-03-14T14:25:54</dateUpdated>
<experimenter id="ID0001">
<firstName>Jamien</firstName>
<lastName>Hoebeeck</lastName>
<labName>Center for Medical Genetics Ghent</labName>
</experimenter>
<dye id="SYBRGreen I" />
<sample id="gDNA">
<type>unkn</type>
</sample>
[...]
<target id="Exon 1">
<description>von Hippel-Lindau tumor suppressor - Exon 1 set 2</description>
<xRef>
<name>symbol</name>
<id>VHL</id>
</xRef>
<xRef>
<name>rtprimerDb</name>
<id>1023</id>
</xRef>
<xRef>
<name>entrezId</name>
<id>7428</id>
</xRef>
<type>toi</type>
<amplificationEfficiency>0.95</amplificationEfficiency>
<meltingTemperature>87.7</meltingTemperature>
<dyeId id="SYBRGreen I" />
<sequences>
<forwardPrimer>
<sequence>CGCCGCATCCACAGCTA</sequence>
</forwardPrimer>
<reversePrimer>
<sequence>GGCTTCAGACCGTGCTATCG</sequence>
</reversePrimer>
<amplicon>
<sequence>CGCCGCATCCACAGCTAccgaggtacgggcccggcgcttaggcccgacccagcagggaCGATAGCACGGTCTGAAGCC</sequence>
</amplicon>
</sequences>
</target>
[...]
<thermalCyclingConditions id="PCR10350">[...]</thermalCyclingConditions>
<experiment id="Experiment_1">
<description>Experiment_description</description>
<run id="Run_1">
<pcrFormat>
<rows>8</rows>
<columns>12</columns>
<rowLabel>ABC</rowLabel>
<columnLabel>123</columnLabel>
</pcrFormat>
<react id="1">
<sample id="gDNA" />
<data>
<tar id="Exon 1" />
<cq>23.2</cq>
<adp>
<cyc>3</cyc>
<fluor>668.43</fluor>
</adp>
[...]
</data>
</react>
</run>
</experiment>
</rdml>As adoption was underwhelming, a simplified sister format was conceived. The Real-time PCR Data Essential Spreadsheet Format (RDES) is a character-separated text file that can be worked on with spreadsheet software.
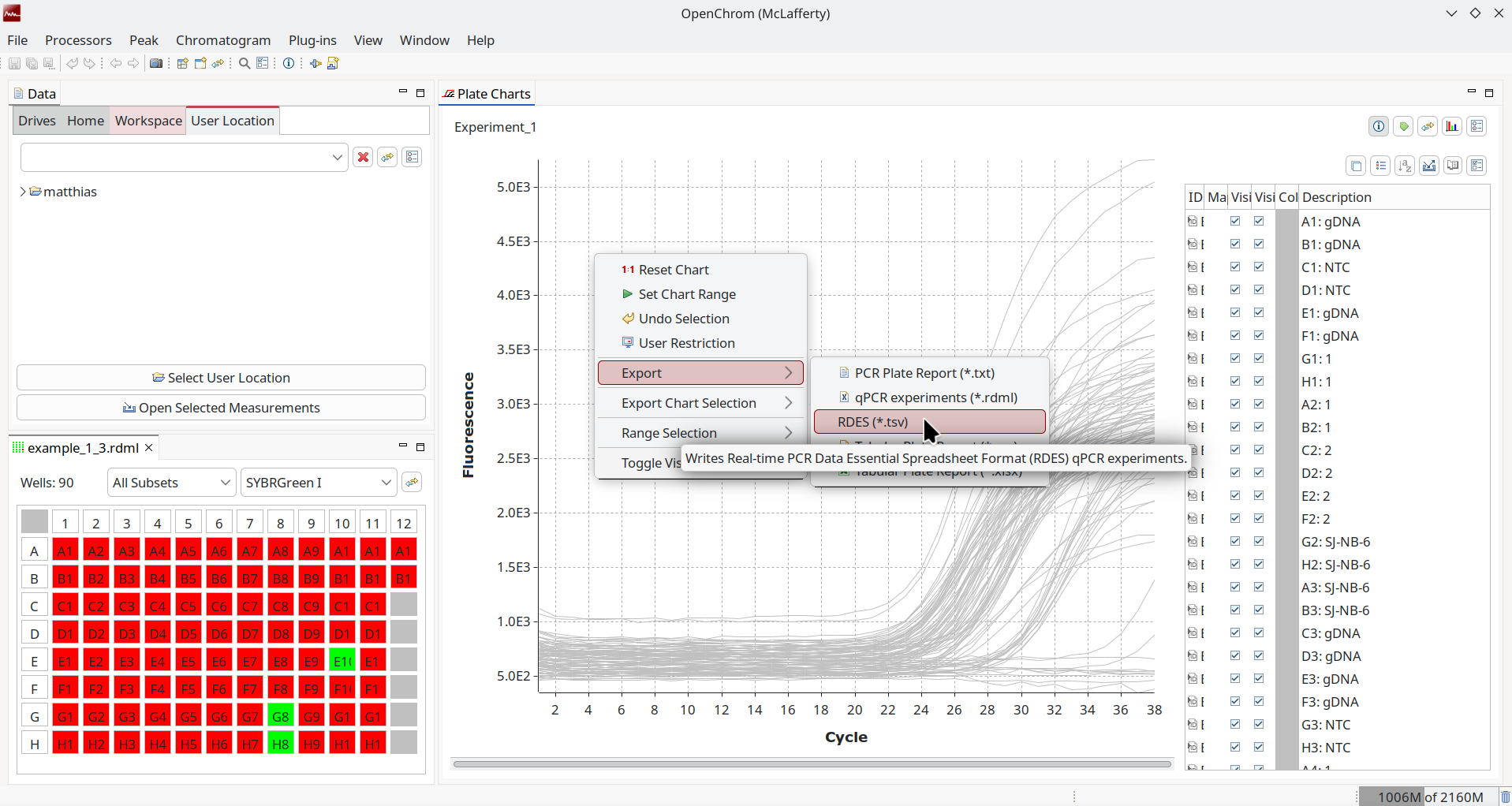
It may be a surprise, but OpenChrom also has support for real-time qPCR data and also adopted the proposed open formats with read/write support for RDML and write support for RDES. So even if this format has been neglected by vendors of PCR cyclers, you can use OpenChrom to convert to FAIR data.
mzDB
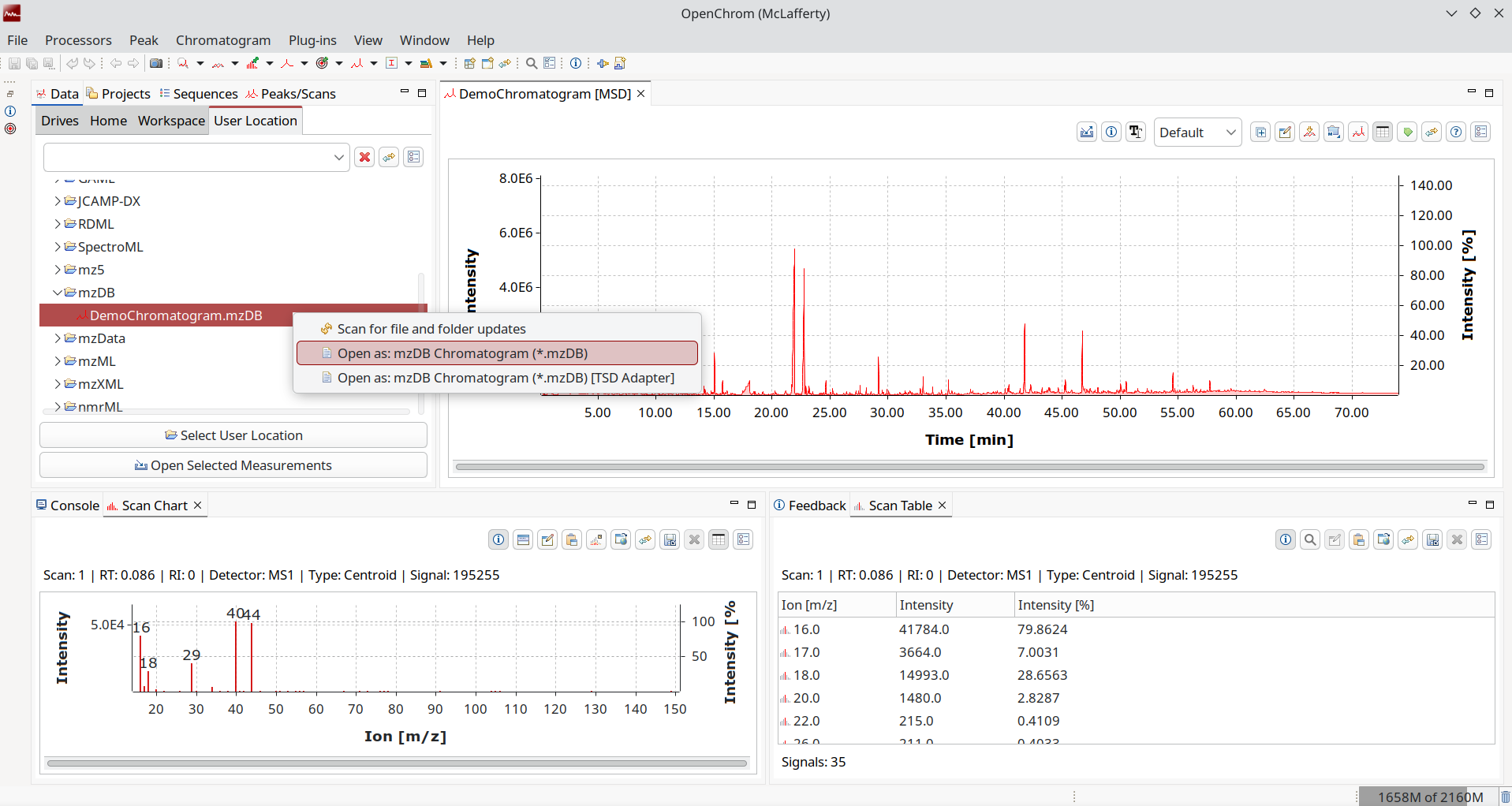
Again, we have mass spectrometric data but a new take. What if we save our tandem MS chromatograms in a database to disk? This way we can query it faster! Excellent for large files where you are only interested in selected smaller bits from mass spectrometric techniques that create a lot of raw data per run.
You can use existing tools like SQLiteBrowser to explore the files. OpenChrom has read/write support for this novel approach, even though it might be a bit niche.
mz5/mzMLb
mzML is great, but it does not scale for larger files. By using an HDF5 standardized binary container, we can increase time/speed efficiency, as in faster read/write speeds and better compression. To not reinvent the wheel, mzMLb saves the XML in binary form and splits the data, while mz5 reuses the fields but saves them in HDF5 native types. If you already worked with mzML you will feel right at home.
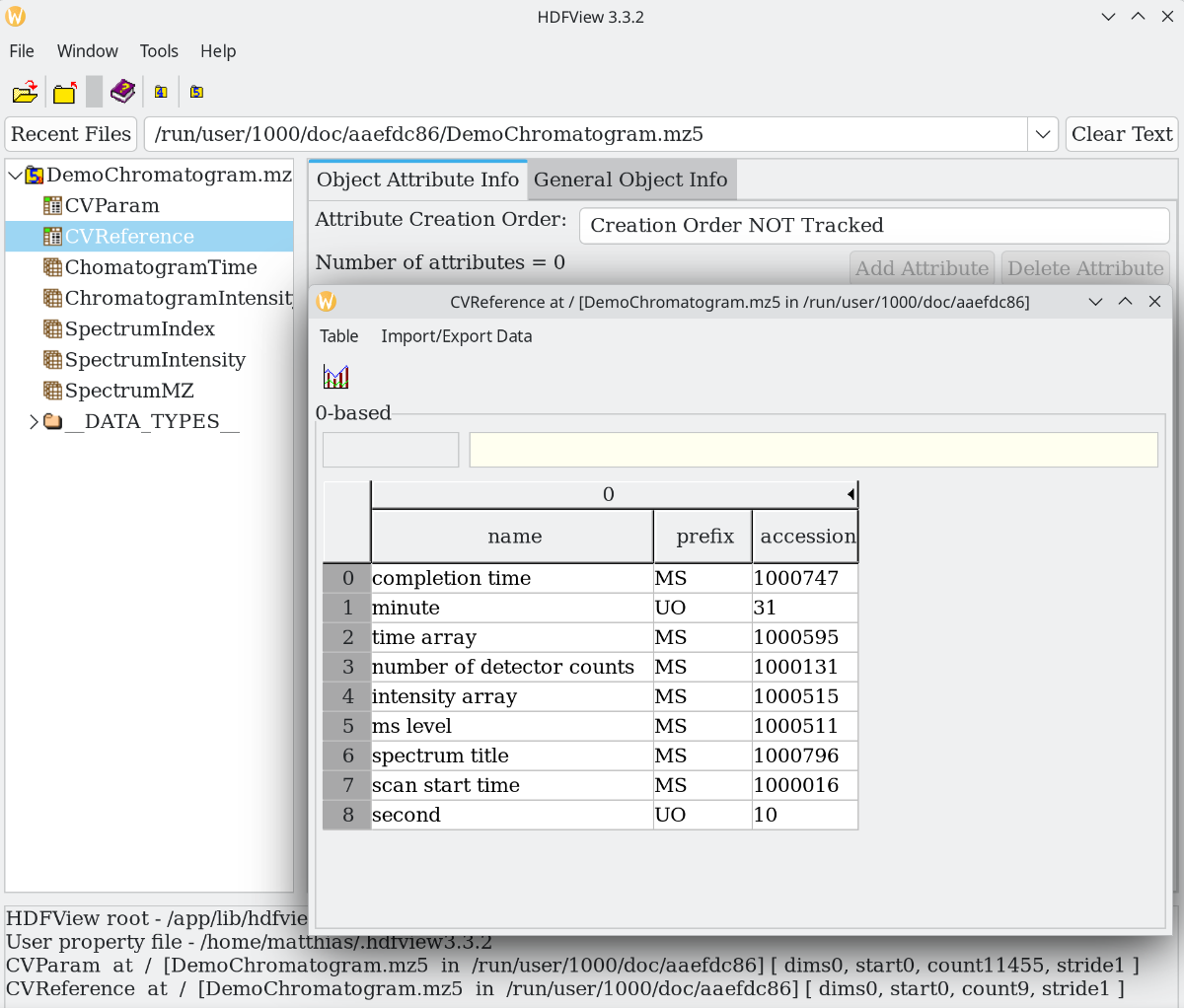
Like netCDF, we can explore this with a generic viewer like HDFview because of its open and self-describing nature.
Just like mzML, we support read/write with OpenChrom and highly recommend it in case you are bottlenecked by existing formats and need something compatible with established proteomics pipelines.
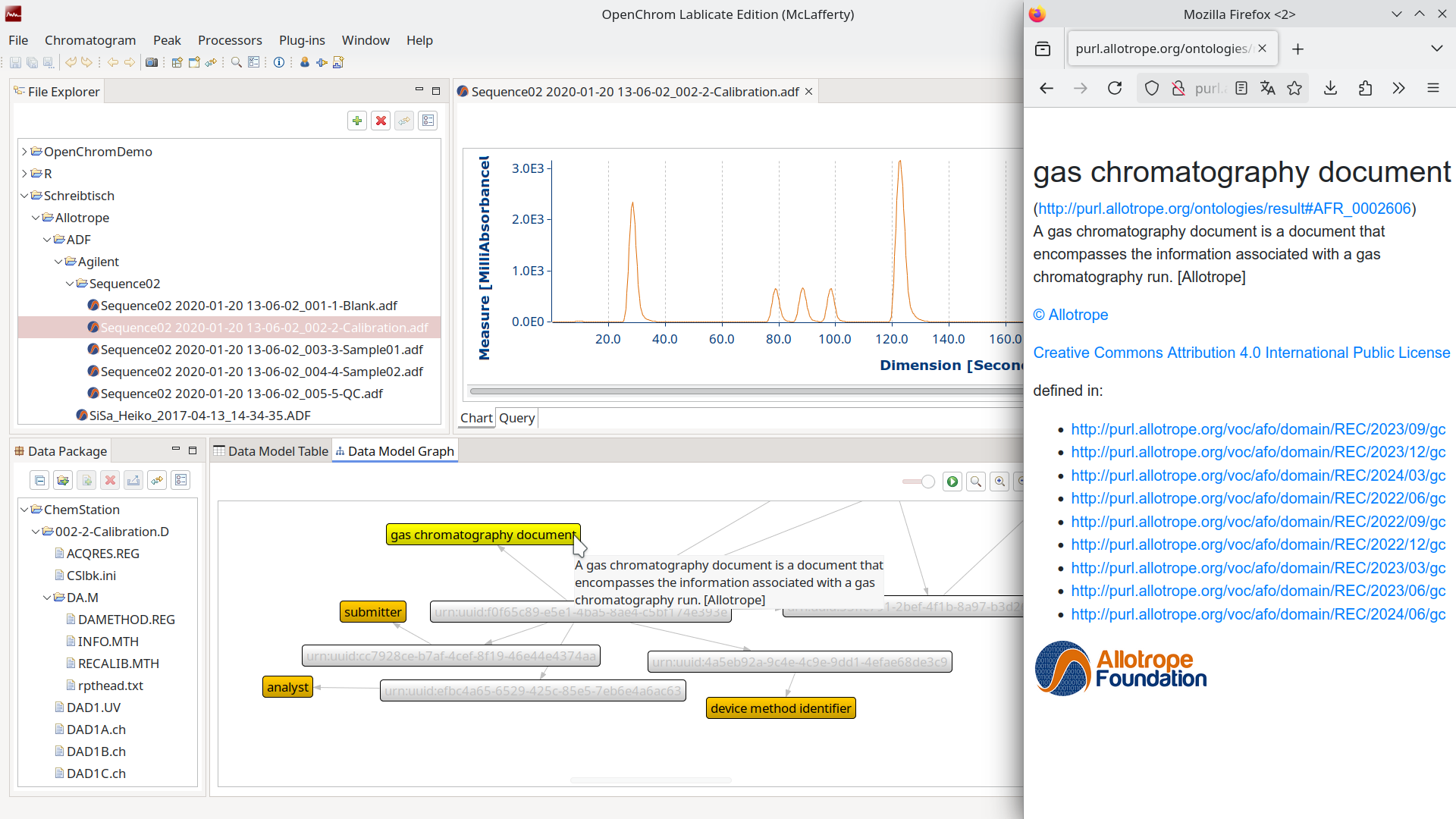
Allotrope Data Format/Simple Model

The Allotrope Data Format (short ADF) is again an HDF5 container, but it encodes the metadata in a W3C Triplestore using the RDF Data Cube Vocabulary. This is another web technology in the form of subject, predicate, and objects to map relations. The standard is governed by the Allotrope Foundation, which also publishes an ontology for use within the file formats. There is a membership fee and a library to work with the files. So it is an open standard with a self-describing format but behind a paywall, and you inherit all the technical decisions that inventor OSTHUS made in the past.
To increase adoption, the Allotrope Foundation conceived another standard that uses their ontology, but this time it is JSON-based. The Allotrope Simple Model (known as ASM) is basically structured text that is a lot easier to read and write with existing tooling. The schemas for analytical techniques can be used to validate and are curated by the foundation with quarterly releases under a Creative Commons license. You need to be a member to suggest changes or draft new ones. It is organized more like an open-source project, but it is still a members-only club.
OpenChrom has read/write support for both and offers specialized tooling to inspect any files that adhere to the standard. You can read about both ADF/ASM in a previous blog post where we covered it extensively: